
Mol Cell: 超级增强子突变促黑色素瘤生长
BET,溴结构域和超末端结构域家族,它的抑制剂BETi目前是很有前景的转移性黑色素瘤治疗药物,但作用机制尚不明确。最近,来自纽约大学郎格尼医学中心的研究者于Molecular Cell 发表了一篇文章,解答了这个问题。
1. AMIGO2—黑色素瘤的生存基因
研究者从对BET抑制剂JQ1敏感的基因中,逐步筛选出黑色素瘤细胞特异的78个过表达的原致癌基因,再从中选择出8个尚未报道与黑色素瘤发生发展有关的基因,接受功能缺失性筛选:
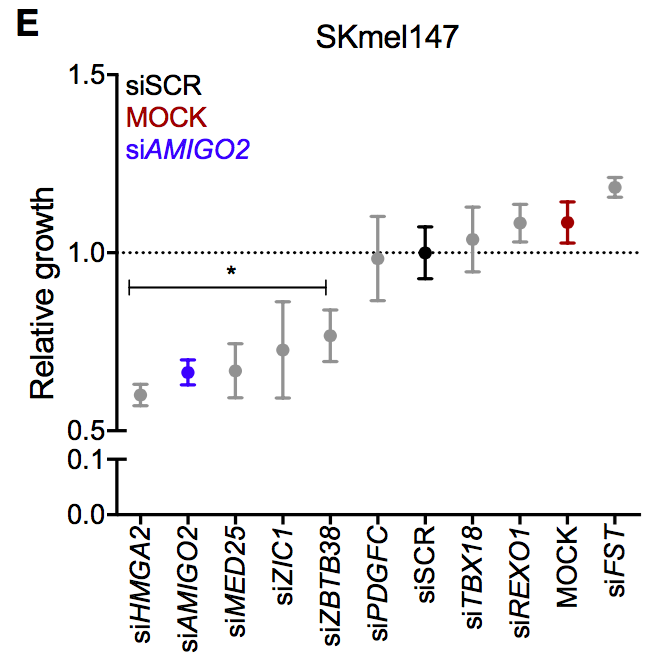
siRNA实验结果显示,除了阳性对照HMGA2,令黑色素瘤细胞SKmel147增殖缺陷最明显的是AMIGO2。
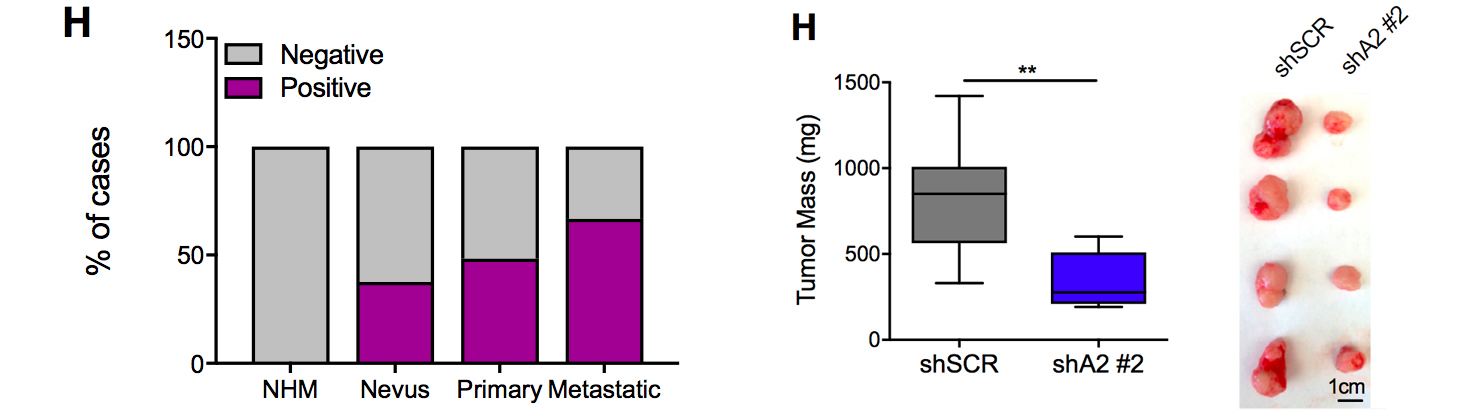
左图:黑色素瘤的AMIGO2 蛋白质水平比正常黑色素细胞NHM高;右图,敲降AMIGO2,肿瘤生长减缓。
2. 超级增强子的鉴定
已有报道表明,癌基因的表达以及对BETi的敏感性和BET上的超级增强子 (SEs) 以及启动子相关。因此,为了进一步研究AMIGO2正调控黑色素瘤的机制,研究者对SKmel147细胞进行了BRD4的ChIP-seq:
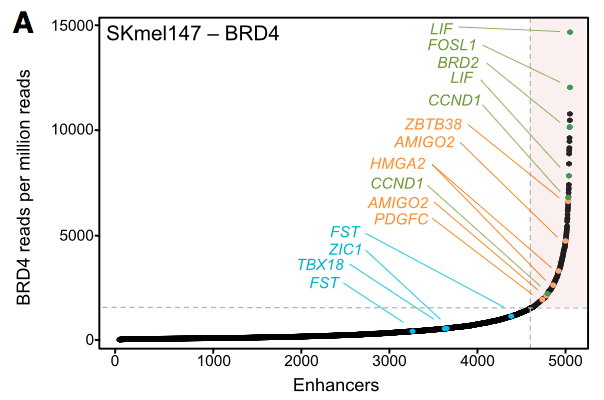
通过BRD4的ChIP-seq,鉴定出SKmel147细胞的464个SEs,4678个TEs,它们同时富集有BRD2、H3K27ac修饰和MED1。其中AMIGO2与两个SEs有关。
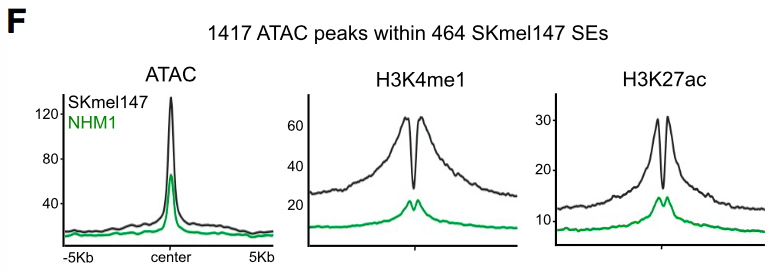
通过ATAC-seq (转座酶可进入性染色质测序) 和H3K4me1、H3K27ac的ChIP-seq,研究者鉴定得黑色素瘤细胞的SEs内有1400个组成型增强子 (constituent enhancers, 组成SEs的普通增强子),这些增强子有着开放的染色质结构、H3K27ac和H3K4me1修饰,但它们在正常黑色素细胞中并没有开放结构和这两种组蛋白修饰,表明正常黑色素细胞癌化涉及增强子的重新分布,从而促进黑色素瘤基因表达。
在SKmel147细胞的AMIGO2 TSS上鉴定得两个突变的SEs,三者位于同一个突变的TAD (拓扑相关结构域) 和LAD (板层关联结构域):

研究者关注更靠近AMIGO2上游的一个SE,它在几种黑色素细胞中都出现,表明AMIGO2受它调控是保守的。
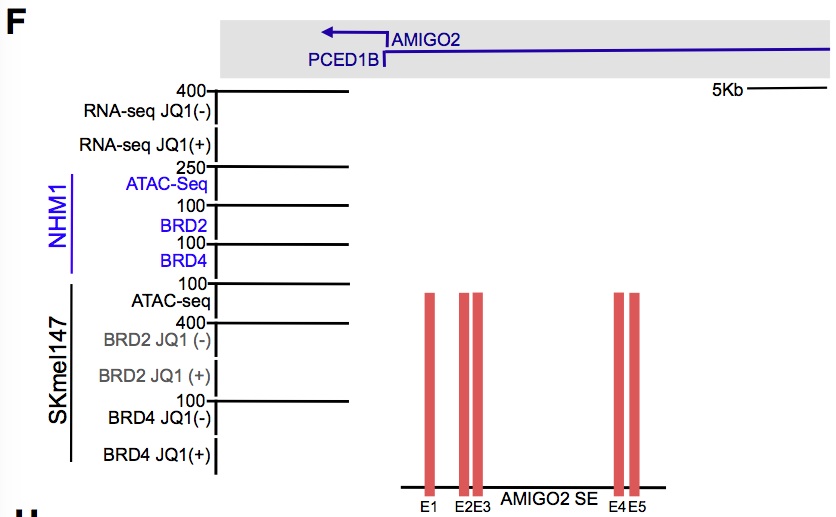
ATAC-seq和ChIP-seq结果表明,在这个SE里有5个组成型增强子 (E1-E5),E1~3在几种黑色素瘤细胞中都有,E4、E5主要在SKmel147细胞中;黑色素瘤细胞的AMIGO2和SEs比正常细胞富集有更多的BRD2、BRD4。
3. 进一步研究机制: SEs、BET、AMIGO2如何联合作用
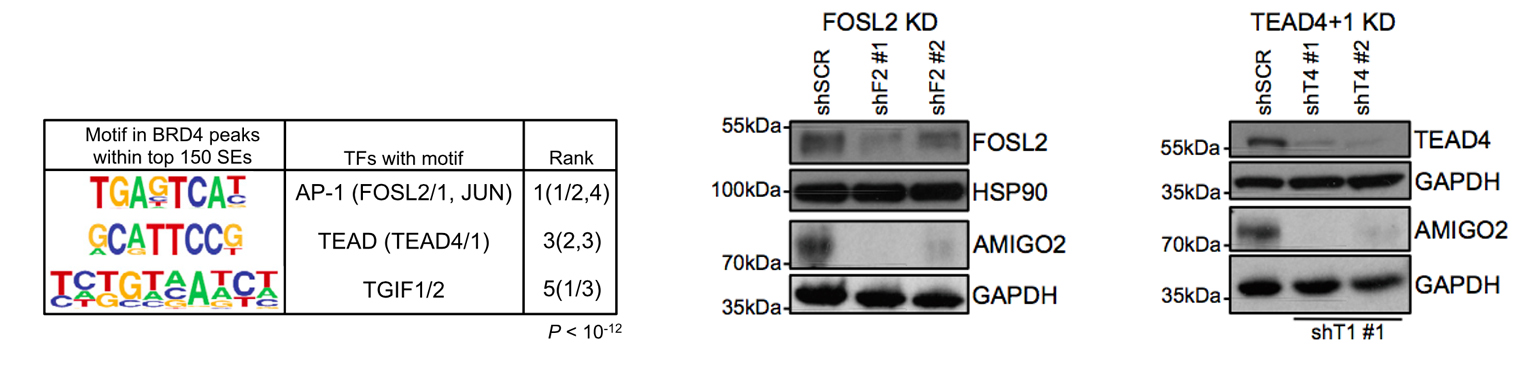
左图,对top 150 SEs进行motif分析,得到FOSL2、TEAD4等TF (转录因子);右图,敲降这两个转录因子,会导致AMIGO2减少,表明AMIGO2在黑色素瘤中的表达与这些TF有关。
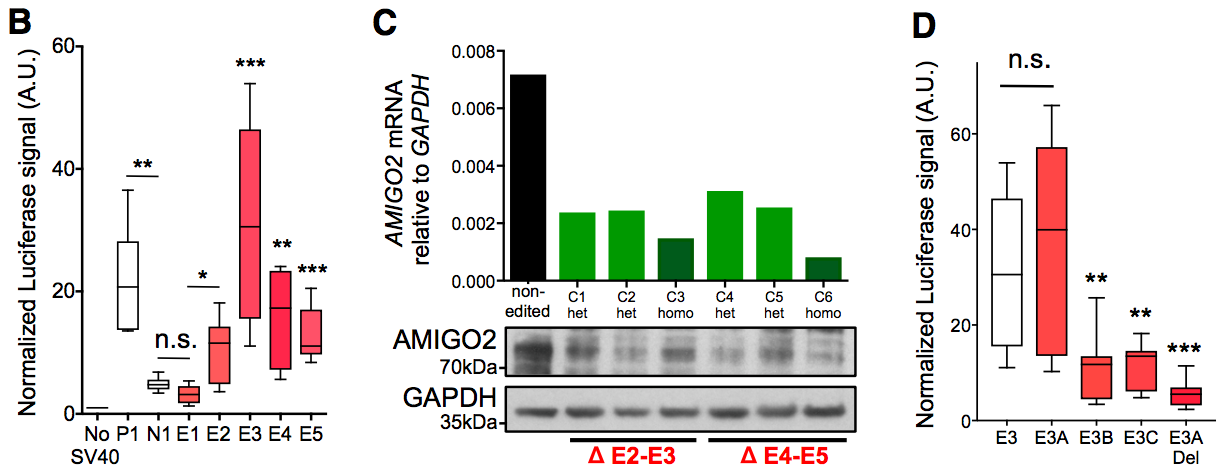
通过启动子荧光素酶表达实验、CRISPR/Cas9删除E2-5实验,发现在黑色素瘤中,更靠近上游的这个SE能调控AMIGO2表达。
4. 总结
这项研究观察到黑色素瘤的增强子发生突变,AMIGO2 SE存在于所有研究的黑色素瘤细胞类型中,但是在正常的黑色素细胞中没有活性。研究者认为,在转化的过程中,这个SE是具有活性的,然后获得BRD2、BRD4、FOSL和TEAD转录因子;在BETi处理后,BET从AMIGO2 SE和启动子上离开,导致转录下调。但是仍不清楚BETi能否让这些TF脱位,以及BET蛋白和TF是怎样与AMIGO2 SE合作的,这些疑点有待研究者们继续探索。
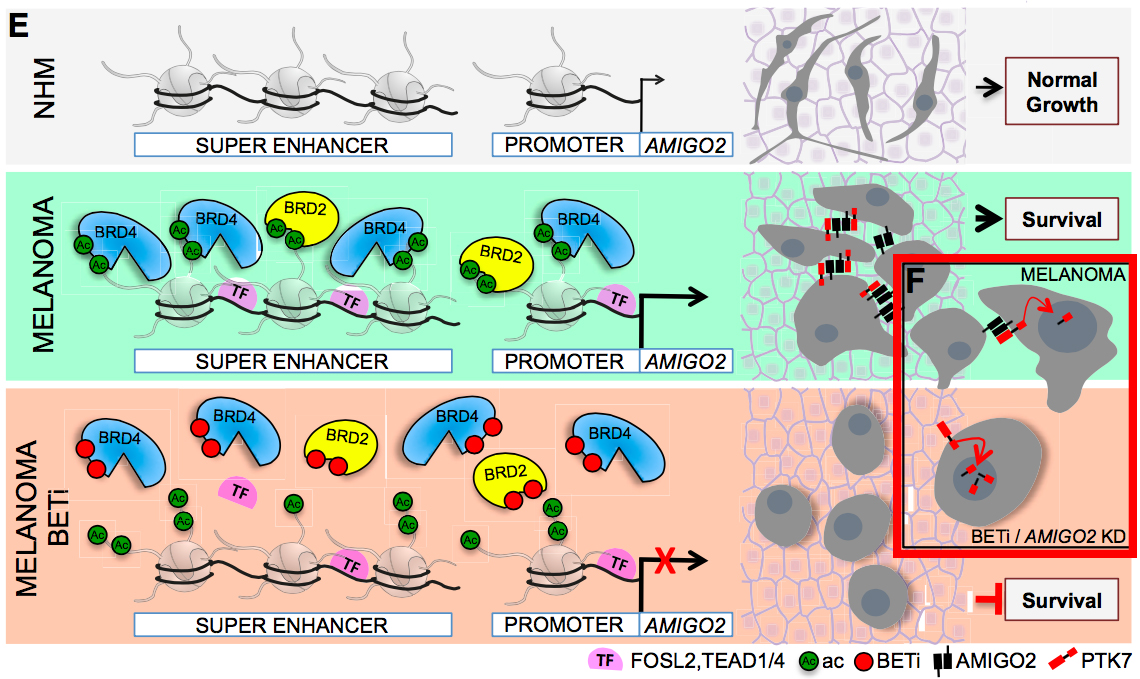
E.正常黑色素细胞中的AMIGO2表达水平很低,且没有活性SE;癌化的时候,BRD2和BRD4增加,转录因子FPSL2和TEAD4/1发生变化,引发全范围的表观重编程,包括产生SE,激活关键黑色素瘤生存基因,如AMIGO2;BETi处理后,BET蛋白从SE和启动子上离开,使基因沉默。F. AMIGO2与PTK7相互作用,抑制CTF2-PTK7在核累积。在BETi和AMIGO2 KD的作用下,CTF2-PTK7核水平上升,可能导致基因表达的变化。
原文:Fontanals-Cirera B, et al. Harnessing BET Inhibitor Sensitivity Reveals AMIGO2 as a Melanoma Survival Gene. Mol Cell. 2017 Nov 16;68(4):731-744.e9.
- - - 推荐阅读 - - -
振奋人心的3D基因组技术:GRID-seq探索RNA-DNA相互作用
突破性进展!UCSD付向东团队开发新技术,首次实现全基因组范围RNA-DNA相互作用的全谱鉴定!
2017-12-01
lncRNA研究策略—来自Science一作王品博士的经验分享
表观生物公众号很荣幸邀请到了王博士为广大的 lncRNA研究者分享了他对非编码RNA的研究心得和策略,大家一起来学习交流吧!
2017-11-21

这篇 Cell 文章告诉你如何研究外泌体 miRNA
研究外泌体miRNA的文章越来越多,可是高分的并不多。怎么样的外泌体实验结果在审稿人眼中才具有说服力?Epi老师今天带你们看看这篇最近发表于Cell的外泌体miRNA文章。 肥胖引起的慢性组织炎症是胰岛
2017-11-02