
Pacbio 全长转录组 Iso-seq 服务
展示与解读1:Iso-Seq基本分析
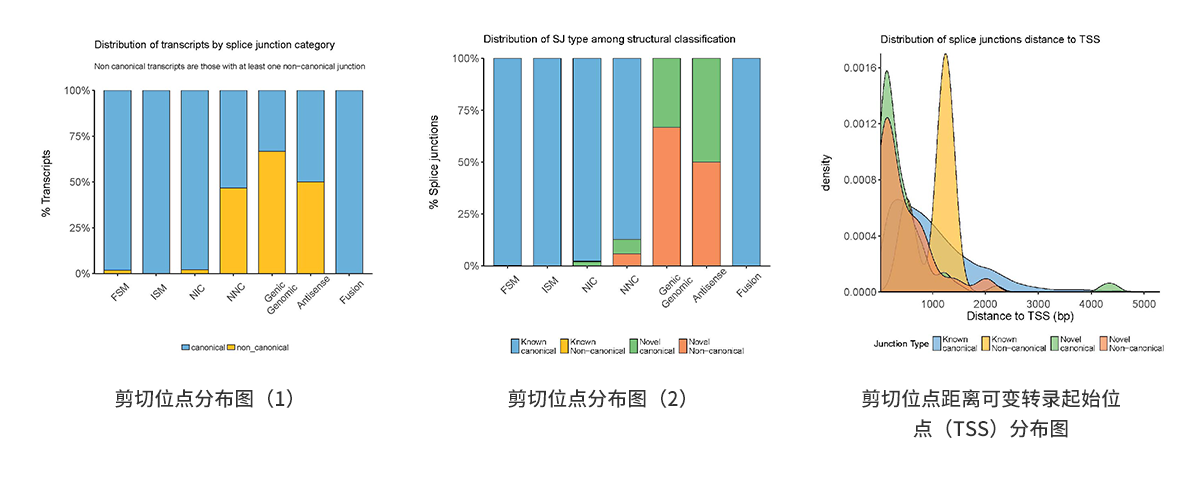
(7)剪切位点分析
可变剪切(Alternative Splicing, AS),是大多数真核生物细胞中普遍的一种基因表达方式。真核细胞的基因序列包含内含子(intron)与外显子(exon),在基因转录成 mRNA 前体后内含子会被 RNA 剪切体移除,而外显子则保留于成熟 mRNA 中。在一条未经剪切的前体RNA中,可以具有多种外显子剪切形式,因此使得一个基因在不同时间、不同环境中可以翻译出不同的蛋白质,进而增加其生理状况下系统的复杂性或适应性。剪切位点的分析可以帮助我们找到新的剪切方式。
参考文献
[1] Manuel, Tardaguila, Lorena, et al. Corrigendum: SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification.[J]. Genome Research, 2018.
[2] Tian L, Shao Y, Nance S, et al. Long-read sequencing unveils IGH-DUX4 translocation into the silenced IGH allele in B-cell acute lymphoblastic leukemia. Nat Commun. 2019;10(1):2789. Published 2019 Jun 26. doi:10.1038/s41467-019-10637-8
[3] de la Fuente, L., et al. (2019). "tappAS: a comprehensive computational framework for the analysis of the functional impact of differential splicing." 690743.
[4] Li, H. (2017). "Minimap2: fast pairwise alignment for long DNA sequences."

展示与解读2:Iso-Seq高级定制分析
(7) 转录水平功能机制分析
DPA分析使得对3'UTR区域调控机制的差异化显示出来,进而对该区域进行转录本水平的注释,将与富集分析结果共同揭示转录后调控机制,转录水平机制分析如下:

展示与解读2:Iso-Seq高级定制分析
(6) polyA差异表达分析(DPA)
在生物体内,mRNA的3'UTR区域存在着多种调节元件,而UTR的长度变化和可选择性多聚腺苷酸化(alternative polyadenylation,APA)的差异能够影响到mRNA的稳定性,翻译效率和RNA蛋白结合等问题,DPA分析将APA和UTR长度变化结合来揭示转录后调控的机制,DPA分析如下图:

展示与解读2:Iso-Seq高级定制分析
(5) 特征差异表达性分析(DFI)
在生物体内,mRNA上会结合多种功能元件,进而调节转录后的表达水平,研究这些功能元件在异构体和AltTP中的差异变化机制,是揭示转录后调控机制的重要手段,DFI分析是在异构体中研究功能元件和AltTP差异变化的分析内容,如下图所示:

展示与解读2:Iso-Seq高级定制分析
(4) 富集分析
为了研究生物体内AltTP对基因表达调控的功能性影响,对DE和DIU分析后的差异表达进行富集功能分析,直接比较两种差异分析机制控制的富集分析,能够找到与转录本调节相关的功能元件和转录控制的触发过程,进而找到具有调节功能的特定亚型,富集分析如下图:
展示与解读2:Iso-Seq高级定制分析
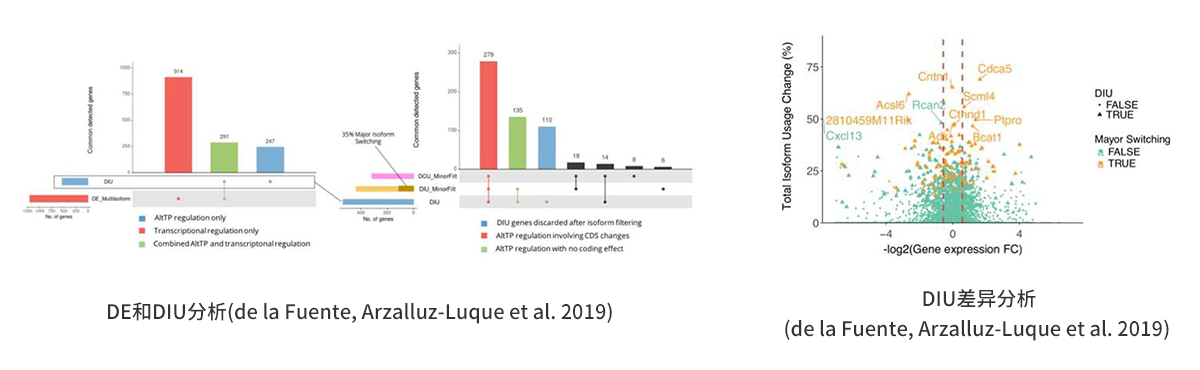
(3) 差异和异构体差异表达性分析(DE/DIU)
转录和转录后调控控制总表达水平的变化,转录本差异表达、基因差异表达和CDS差异表达的分析称为DE差异表达分析,同时转录本异构体差异表达分析称为DIU差异表达分析,结合DE和DIU分析,能够发现转录和转录后调控表达水平的变化,如下图:
展示与解读2:Iso-Seq高级定制分析
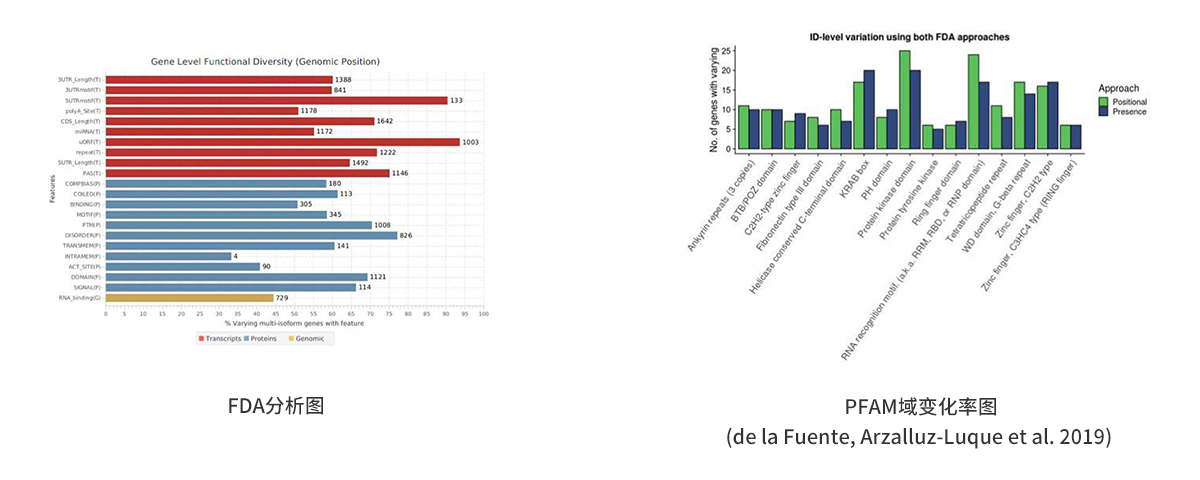
(2) 功能多样性分析(FDA)
在生物体内,转录后调控机制实现了生物体内的复杂功能,研究转录后调控机制很大程度上取决于转录亚型是否包含功能元件的变异,它们的表达水平的改变可以有效的调节生物体功能的变化,功能多样性分析(FDA)是定量这些功能元件的变化,为找到差异性功能元件而设计的分析过程,FDA分析,结果如下图:

展示与解读2:Iso-Seq高级定制分析
(1) AltTP分析
AltTP(Alternative Transcript Processing)(de la Fuente, Arzalluz-Luque et al. 2019),是可变剪切位点(AS)、可变聚腺苷酸化位点(APA)和可变转录起始位点(TSS)三者的总称,我们将同时在一条转录本上分析这3个位点的差异和功能,是揭开转录后调控机制的重要手段。

技术原理
Pacbio 全长转录组 Iso-seq 服务
全长转录组测序(Iso-Seq)是基于PacBio Sequl II的单分子实时测序技术。该平台凭借超长读长的优势,无需打断RNA分子,直接对反转录的全长cDNA测序,即可得到从5'末端到3'PolyA尾的高质量全长转录本序列,从而准确鉴定异构体,可实现对可变剪切(Alternative Splicing, AS)、可选择性多聚腺苷酸化(alternative polyadenylation,APA)、
可变转录起始位点(Alternative Transcription Start Sites ,ATSS)等多项二代测序所无法实现的分析能力,打开了一扇通往RNA世界的新大门!
技术优势: 1. 三代测序无需组装可获得准确度大于99%的高质量转录本(HQ high-quality isoforms) 2. 使用最新Iso-Seq3分析流程,获取最新分析手段 3. 使用最新的全长转录本的功能分析(FIT Functional Iso-Transcriptomics analysis) 4. 数据分析多种选择,定制分析内容
展示与解读1:Iso-Seq基本分析
(6)融合基因鉴定
融合基因是指将两个或多个基因的编码区首尾相连.置于同一套调控序列(包括启动子、增强子、核糖体结合序列、终止子等)控制之下,构成的嵌合基因。融合基因通过染色体变异形成的,包括染色体易位、倒置、大片段缺失和插入等。自从发现周期性融合基因 BCR-ABL1 表达将导致慢性髓细胞性白血病(CML)以来,越来越多的肿瘤的发生都与基因融合相关。因此检测融合基因对应研究肿瘤发生有重要意义。
展示与解读1:Iso-Seq基本分析
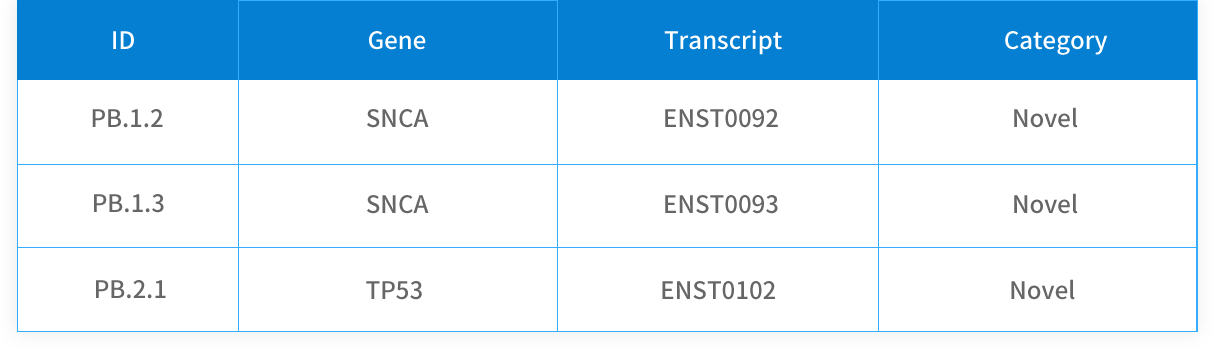
(5)新转录本预测
根据注释类型,NIC和NNC类型转录本即为新转录本,统计如下:

展示与解读1:Iso-Seq基本分析
(4)转录本结构注释
将得到的全长非冗余转录本序列通过 minimap2 软件比对到参考基因组上,进行转录本结构注释。根据转录本和参考注释信息比对,得到如下注释类型:
FSM: 完全匹配上参考序列的转录本
ISM:部分匹配上参考序列的转录本
NIC: 新型转录本亚型,与参考序列有部分外显子重叠
NNC:新型转录本亚型,至少有一个新型剪切位点
Genic Intron: 内含子序列
Genic Genomic: 部分匹配到外显子和内含子区域序列

展示与解读1:Iso-Seq基本分析
(3) 全长转录本去冗余
由于全长转录本在聚类过程中会发生同一转录本分配到不同cluster中,以及3'端不同长度的polyA,5'端差异同样可能导致同一转录本的分配位置差异,使冗余序列产生。
展示与解读1:Iso-Seq基本分析
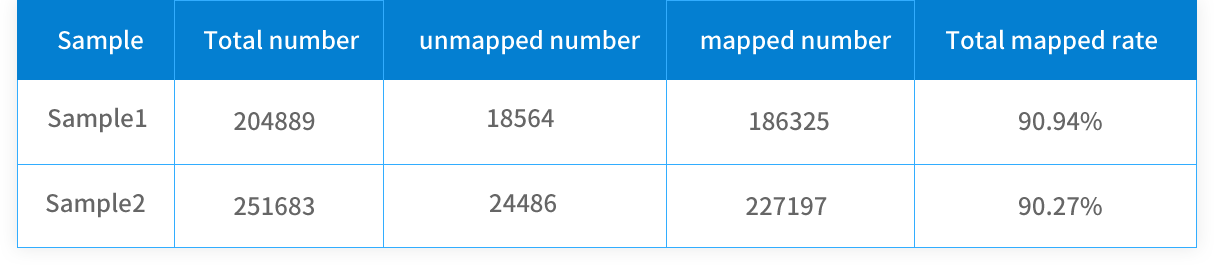
(2)全长转录本与基因组比对
对于整合的全长转录本,我们与相应的参考基因组比对(Li 2017),获得比对结果如下表:
展示与解读1:Iso-Seq基本分析
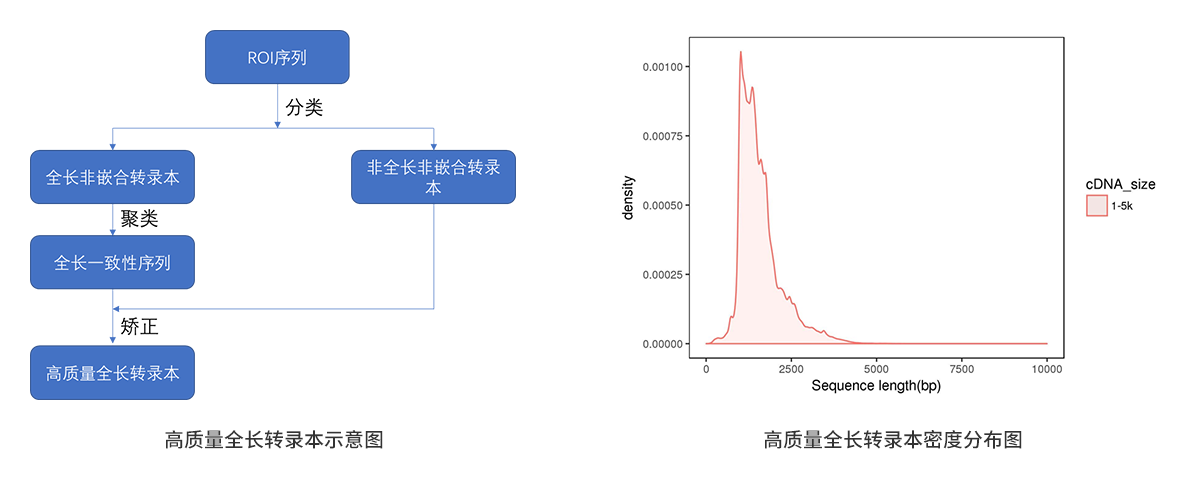
(1) 获得高质量全长转录本示意图:
我们将质控过后的数据ROI,按照是否含有 3´引物和 5´引物,及 3´引物前是否含有 polyA 尾将 ROI 分为全长转录本和非全长转录本;按照序列内部有无测序引物将 ROI 分为嵌合转录本和非嵌合转录本;对 ROI 分类后,取全长非嵌合的转录本进行聚类,去掉一部分冗余的转录本;为了提高序列的准确性,聚类后的全长非嵌合转录本可以用非全长的转录本进行序列矫正,最终获得高质量的全长转录本和低质量的全长转录本。详细的分析流程如下左图:
通常情况下,全长转录本长度分布和数目可以反应cDNA 文库构建的情况以及测序质量的好坏。ROI分类后的全长转录本长度分布应与与建库大小相一致,片段越长的文库获得的全长转录本就越长。此外,全长转录本占 ROI 的比例与文库类型也有关,通常情况下,1-2k 文库中所占的比例在 50%-70%,3-5k 文库中所占的比例在 30%左右。
分析内容
Iso-Seq基本分析
1、 获得高质量全长转录本
2、 全长转录本与基因组比
3、 全长转录本去冗余
4、 转录本结构注释
5、 新转录本预测
6、 融合基因鉴定
7、 剪切位点分析
Iso-Seq高级定制分析
1、AltTP分析
2、功能多样性分析(FDA)
3、差异和异构体差异表达性分析(DE/DIU)
4、富集分析
5、特征差异表达性分析(DFI)
6、polyA差异表达分析(DPA)
7、转录水平功能机制分析
样本要求
5*10^6~1*10^7
细胞数量
10mg
组织质量
20ug
Total RNA